Glycosaminoglycans (GAGs) are critical components of the stem cell niche and consist of long chain polymers of recurring disaccharide units usually composed of either D-glucosamine or D-galactosamine, and D-glucuronic acid or L-iduronic acid that when coupled to a core protein result in the formation of proteoglycans (PGs). Perhaps the most widely used of the GAG-based drugs is the highly sulfated anticoagulant heparin, which is used during surgery and dialysis, as a treatment of deep-vein thrombosis, and is used to coat indwelling catheters and other devices where the vascular system is exposed. Heparin is isolated from animal tissue, primarily pig intestines although it can be available from bovine and ovine intestines and other organs. Due to the animal source, the early stages of heparin production, e.g., generation of the raw heparin, occurs under poorly controlled, non-cGMP conditions, and purposeful contamination with related polysulfated polysaccharides was determined to result in the death of over 80 people in the U.S. in 2008. This has prompted the call for a non-animal sourced heparin.
In collaboration with the late Prof. Robert Linhardt, we have developed a biomanufacturing approach to a bioengineered heparin (Figure 1). Using the E. coli capsular polysaccharide heparosan, a series of chemical and enzymatic transformations are being optimized to generate a chemically and biologically equivalent (to USP) heparin. We have gained an improved understanding of the various sulfotransferases in the biosynthesis of heparin. Specifically, we developed a high-throughput microtiter-based colorimetric assay for elucidating the kinetic mechanism of heparin O-sulfotransferases (OSTs) acting on polysaccharide substrates using a given OST with arylsulfotransferase-IV and p-nitrophenylsulfate as sacrificial sulfate donor to regenerate the PAPS OST substrate.
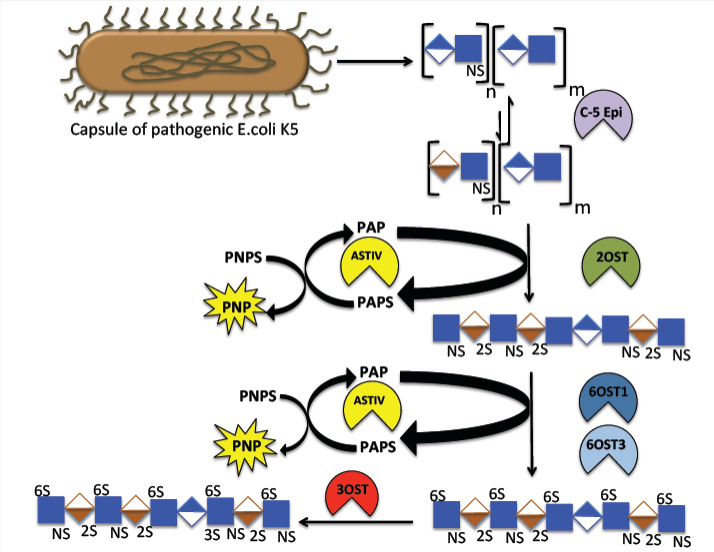
Figure 1. Biosynthetic pathway of heparin. The biosynthetic pathway includes the biosynthesis of polysaccharide backbone as well as the modification steps. The synthesis is initiated with a tetrasaccharide linkage region that contains xylose-galactose-galactose-glucuronic acid. The backbone is synthesized by HS polymerase. The backbone polysaccharide is then modified via five enzymatic modification steps. Relevant References: Bhaskar et al. Angew. Chem. Int. Ed. 58, 5962-5966 (2019); Zhang et al. Anal. Bioanal. Chem. 401, 2793-2803 (2011); Vaidyanathan et al. Bioeng Transl Med. 2, 17-30 (2017); Bhaskar et al. Appl. Microbiol. Biotechnol. 93, 1-16 (2012); Wang et al. Bioengin. Bugs 2, 1-5 (2011); Wang et al. Biotechnol. Bioeng. 107, 964-973 (2010); Ly et al. Anal. Bioanal. Chem. 399, 737-745 (2011); Hickey et al. J. Biotechnol. 165, 175-177 (2013); Bhaskar et al. Carbohydr. Polym. 122, 399-407 (2014); Fu et al. J. Med. Chem. 60, 8673-8679 (2017).
We have focused on both applied and fundamental research on heparin biosynthesis. With respect to the former, we have developed the first bioengineered heparin (BEH) with chemical, biological, and pharmacological equivalence to porcine intestinal heparin, the current FDA approved heparin. Heparins have been invaluable therapeutic anticoagulant polysaccharides for over a century, whether used as unfractionated heparin (UFH) or as low molecular weight heparin (LMWH) derivatives. However, heparin production by extraction from animal tissues presents multiple challenges, including the risk of adulteration, contamination, prion and viral impurities, limited supply, insecure supply chain, and significant batch-to-batch variability. The use of animal-derived heparin, currently only sourced from porcine intestinal mucosa in the US, also raises ethical and religious concerns, as well as carries the risk of transmitting zoonotic diseases. Chemoenzymatic synthesis of animal-free heparin products would offer several advantages, including reliable and scalable production processes, improved purity and consistency, and the ability to produce heparin polysaccharides with molecular weight, structural and functional properties equivalent to those in of the United States Pharmacopeia (USP) heparin. We report a scalable process for production of bioengineered heparin that is biologically and compositionally similar to USP heparin (Figures 2 and 3). This process relies on enzymes from the heparin biosynthetic pathway, immobilized on an inert support, and requires a tailored N‑sulfoheparosan with N-sulfo levels similar to those of porcine heparins. We also report the conversion of our bioengineered heparin into a LMWH that is biologically and compositionally equivalent similar to USP enoxaparin. Ultimately, we demonstrate major advances to a process to provide a potential clinical and sustainable alternative to porcine-derived heparin products [Douaisi et al. Proc. Natl. Acad. Sci. USA, 121, e2315586121, 2024].
A similar method was used to generate a low molecular weight heparin (LMWH) [Yu et al. Carbohydrate Polym. 295, 119825, 2022]. LMWH is prepared from the controlled chemical or enzymatic depolymerization of animal sourced heparins. It has been widely used as an anticoagulant. Concerns about the shortcomings of animal-derived heparin and the contamination of supply chain demand biochemical approaches for synthesizing LMWH. In the present study, two LMWHs were enzymatically synthesized from low molecular weight N-sulfated heparosan (LMW-NSH) cleaved by recombinant hydrolase, endo-β-glucuronidase, (HepBp) or heparin lyase III (HepIII), followed by subsequent sulfotransferase modifications (Figure 4). Structural characterization shows that LMWH chains prepared using HepBp had a saturated uronic acid residue at their reducing ends, while chains of LMWH prepared using HepIII had an unsaturated uronic acid residue at their non-reducing end. Both LMWHs had anti-factor Xa and anti-factor IIa activities comparable to enoxaparin. This approach demonstrates that the hydrolase, HepBp, can be used to prepare a new type of LMWH that has no unsaturated uronic acid at its non-reducing end.
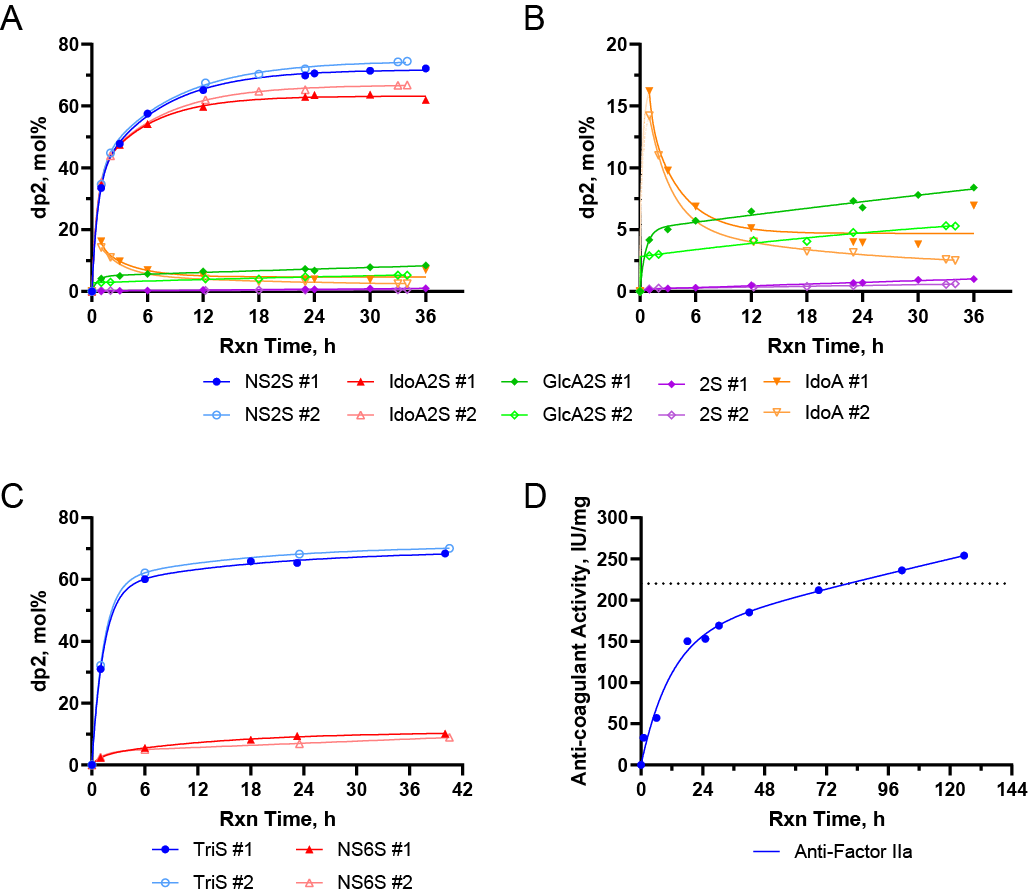
Figure 2. Time courses of multi-gram synthetic reactions leading to a 10 g BEH batch. (A and B) Time course of two sequential sub-batches of the C5-epi/2OST reactions (#1 and #2), with magnification (B) to visualize 2S, IdoA, and GlcA2S disaccharides. (C) Time course of two sequential sub-batches of the 6OST-1/6OST-3 reactions (#1 and #2). (D) Time course of the single batch 3OST-1 reaction. Curves fitted using two-phase association model.
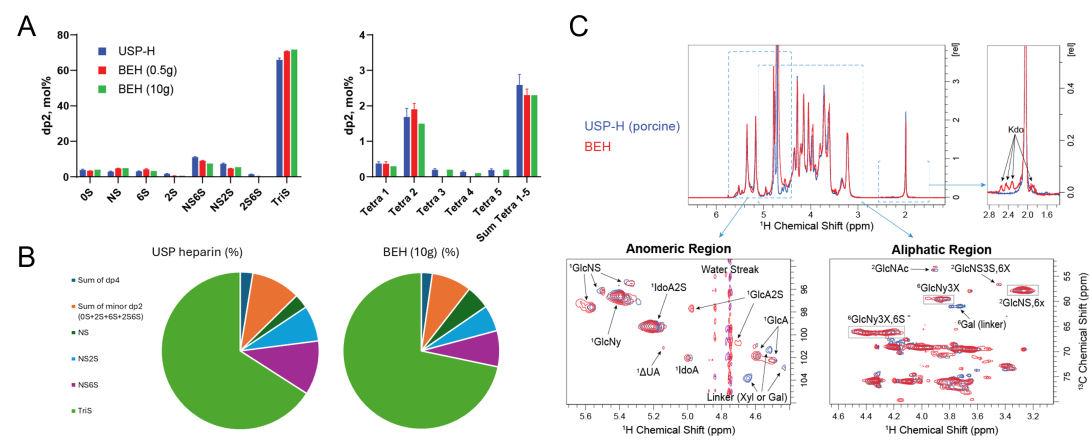
Figure 3. Structural equivalence of bioengineered heparin (BEH) to porcine intestinal USP-heparin. (A) Disaccharide analysis (major components) for USP-heparin and BEH with (B) overall snapshot comparison shown in pie charts. (C) 1D and 2D NMR analysis of USP heparin vs. BEH. BEH (10 g batch) and USP heparin samples were dissolved in 2H2O and lyophilized repeatedly to achieve full deuterium exchange. The samples were then analyzed by 1D 1H and 2D 1H/13C HSQC NMR on 600 MHz spectrometer.
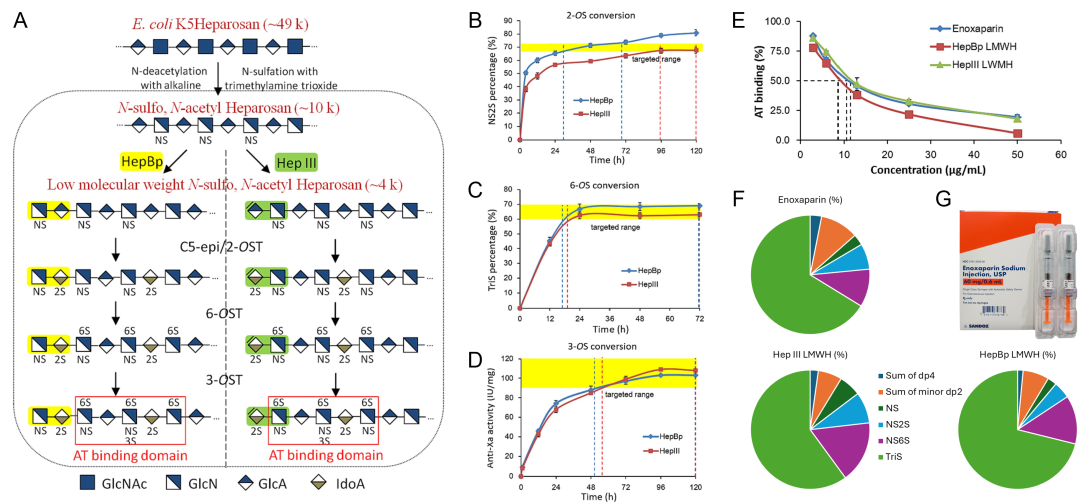
Figure 4. (A) Scheme for enzymatic biosynthesis of LMWH made from HepBp and HepIII cleaved N-sulfated heparosan. (B-D) Sulfotransferase modifications of LMW-NSH. (B) NS2S preparation by conversion of LMW-NSH through C5-epimerase and 2-O-sulfotransferase combined reaction; (C) TriS preparation by conversion of NS2S through 6-O-sulfotransferase reaction; (D) Anti-Xa activity during conversion of TriS to LMWH through 3-O-sulfotransferase reaction. (E) Binding of Enoxaparin and bioengineered LMWH to antithrombin (AT). (F) Overall snapshot comparison of Enoxaparin and bioengineered LMWH composition shown in pie charts. (G) Example of a packaging of Enoxaparin sodium.
With respect to fundamental work, we have examined the mechanism of glucuronosyl C5-epimerase (Glce), the first enzyme that acts on the polysaccharide substrate N-sulfoheparosan (NSH). The chemoenzymatic synthesis of heparin, through a multi-enzyme process, represents a critical challenge in providing a safe and effective substitute for this animal sourced anticoagulant drug. D-Glucuronyl C5-epimerase (C5-epi) is an enzyme acting on a heparin precursor, N-sulfoheparosan, catalyzing the reversible epimerization of D-glucuronic acid (GlcA) to L-iduronic acid (IdoA). Apparent steady-state kinetic parameters for both the forward and the pseudo-reverse reactions of C5-epi have been determined for the first time using polysaccharide substrates directly relevant to the chemoenzymatic synthesis and biosynthesis of heparin [Vaidyanathan et al. Glycobiology, 30, 847-858, 2020] (Figure 5). The forward reaction shows unusual sigmoidal kinetic behavior and the pseudo-reverse reaction displays non-saturating kinetic behavior. The atypical sigmoidal behavior of the forward reaction was probed using a range of buffer additives. Surprisingly, the addition of 25 mM each of CaCl2 and MgCl2 resulted in a forward reaction exhibiting more conventional Michaelis-Menten kinetics. The addition of 2-O-sulfotransferase, the next enzyme involved in heparin synthesis, in the absence of 3’-phosphoadenosine 5’-phosphosulfate (PAPS), also resulted in C5-epi exhibiting a more conventional Michaelis-Menten kinetic behavior in the forward reaction accompanied by a significant increase in apparent Vmax. This study provides critical information for understanding the reaction kinetics of C5-epi, which may result in improved methods for the chemoenzymatic synthesis of bioengineered heparin.
We further elucidated the processibility of C5-epimerase on NSH substrate (Figure 6). Using full length NSH, containing different amounts of N-acetylglucosamine (GlcNAc) residues, we demonstrated that C5-epimerase specificity depends on polysaccharide sequence, particularly the location of GlcNAc residues within the chain [Vaidyanathan et al. Biochemistry, 59, 2576-2584, 2020]. We leveraged deuterium exchange and the novel β-glucuronidase heparanase BP, which cleaves at the GlcA residue to obtain sequence information. Specifically, liquid chromatography-mass spectrometry and gel permeation chromatography of partial/complete heparanase BP digestion products from various NSH substrates treated with C5-epimerase provides information on C5-epimerase activity and action pattern. This study provides insight into optimizing the large-scale production of bioengineered heparin.
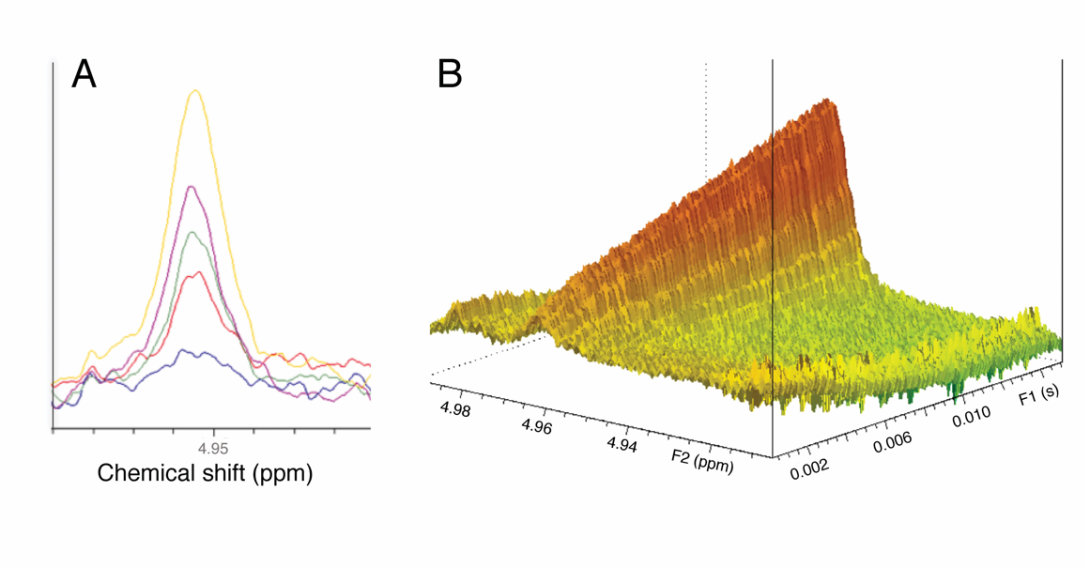
Figure 5. Offline and real-time NMR changes in the H-1 anomeric signal of IdoA produced using C5-epimerase. (A) Spectral overlay using the offline experiments using the engineered enzyme with NSH as the substrate where the IdoA H-1 peak increases as a function of time. The colored lines represent: blue, 15 min; red, 30 min; green, 45 min; purple, 60 min; and yellow, 90 min. (B) Real-time 1D 1H-time spectrum showing an increase in signal corresponding to IdoA H-1 as a function of time (Topspin 3.2.7, contour mode). (C) Michaelis-Menten profiles for engineered C5-epi (0.5 mg/mL), acting on NSH with no additive (black), with 2OST (0.5 mg/mL) added as an additive without PAPS (red) or with added 25 mM CaCl2 + 25 mM MgCl2 (green). Error bars (n = 3) are within the plot symbols. [Vaidyanathan et al. Glycobiology, 30, 847-858, 2020]

Figure 6. Full length N-sulfoheparosan (NSH), a direct substrate for the production of bioengineered heparin containing different amounts of N-acetylglucosamine (GlcNAc) residues, is used as substrate for glucuronyl C5-epimerase. The specificity and action pattern of the enzyme was determined as a function of the polysaccharide sequence and the presence of GlcNAc residues. [Vaidyanathan et al. Biochemistry, 59, 2576-2584, 2020]
Current Collaborators:
Dr. Fuming Zhang, Dr. Marc Douaisi, Dr. Elena Paskaleva – Rensselaer Polytechnic Institute
